PUBLICATIONS
Displaying 27 publications, newest first
No papers found

September 2023 | Research Article

Defining E3 ligase–substrate relationships through multiplex CRISPR screening
Timms RT, Mena EL, Leng Y, Li MZ, Tchasovnikarova IA, Koren I, Elledge SJ
Specificity within the ubiquitin–proteasome system is primarily achieved through E3 ubiquitin ligases, but for many E3s their substrates—and in particular the molecular features (degrons) that they recognize—remain largely unknown. Current approaches for assigning E3s to their cognate substrates are tedious and low throughput. Here we developed a multiplex CRISPR screening platform to assign E3 ligases to their cognate substrates at scale. A proof-of-principle multiplex screen successfully performed ~100 CRISPR screens in a single experiment, refining known C-degron pathways and identifying an additional pathway through which Cul2FEM1B targets C-terminal proline. Further, by identifying substrates for Cul1FBXO38, Cul2APPBP2, Cul3GAN, Cul3KLHL8, Cul3KLHL9/13 and Cul3KLHL15, we demonstrate that the approach is compatible with pools of full-length protein substrates of varying stabilities and, when combined with site-saturation mutagenesis, can assign E3 ligases to their cognate degron motifs. Thus, multiplex CRISPR screening will accelerate our understanding of how specificity is achieved within the ubiquitin–proteasome system.
Specificity within the ubiquitin–proteasome system is primarily achieved through E3 ubiquitin ligases, but for many E3s their substrates—and in particular the molecular features (degrons) that they recognize—remain largely unknown. Current approaches for assigning E3s to their cognate substrates are tedious and low throughput. Here we developed a multiplex CRISPR screening platform to assign E3 ligases to their cognate substrates at scale. A proof-of-principle multiplex screen successfully performed ~100 CRISPR screens in a single experiment, refining known C-degron pathways and identifying an additional pathway through which Cul2FEM1B targets C-terminal proline. Further, by identifying substrates for Cul1FBXO38, Cul2APPBP2, Cul3GAN, Cul3KLHL8, Cul3KLHL9/13 and Cul3KLHL15, we demonstrate that the approach is compatible with pools of full-length protein substrates of varying stabilities and, when combined with site-saturation mutagenesis, can assign E3 ligases to their cognate degron motifs. Thus, multiplex CRISPR screening will accelerate our understanding of how specificity is achieved within the ubiquitin–proteasome system.

September 2023 | Research Article

Elucidation of E3 ubiquitin ligase specificity through proteome-wide internal degron mapping
Zhang Z, Sie B, Chang A, Leng Y, Nardone C, Timms RT, Elledge SJ
The ubiquitin-proteasome system plays a critical role in biology by regulating protein degradation. Despite their importance, precise recognition specificity is known for a few of the 600 E3s. Here, we establish a two-pronged strategy for identifying and mapping critical residues of internal degrons on a proteome-scale in HEK-293T cells. We employ global protein stability profiling combined with machine learning to identify 15,800 peptides likely to contain sequence-dependent degrons. We combine this with scanning mutagenesis to define critical residues for over 5,000 predicted degrons. Focusing on Cullin-RING ligase degrons, we generated mutational fingerprints for 219 degrons and developed DegronID, a computational algorithm enabling the clustering of degron peptides with similar motifs. CRISPR analysis enabled the discovery of E3-degron pairs, of which we uncovered 16 pairs that revealed extensive degron variability and structural determinants. We provide the visualization of these data on the public DegronID data browser as a resource for future exploration.
The ubiquitin-proteasome system plays a critical role in biology by regulating protein degradation. Despite their importance, precise recognition specificity is known for a few of the 600 E3s. Here, we establish a two-pronged strategy for identifying and mapping critical residues of internal degrons on a proteome-scale in HEK-293T cells. We employ global protein stability profiling combined with machine learning to identify 15,800 peptides likely to contain sequence-dependent degrons. We combine this with scanning mutagenesis to define critical residues for over 5,000 predicted degrons. Focusing on Cullin-RING ligase degrons, we generated mutational fingerprints for 219 degrons and developed DegronID, a computational algorithm enabling the clustering of degron peptides with similar motifs. CRISPR analysis enabled the discovery of E3-degron pairs, of which we uncovered 16 pairs that revealed extensive degron variability and structural determinants. We provide the visualization of these data on the public DegronID data browser as a resource for future exploration.

June 2023 | Research Article

Ubiquitin-independent proteasomal degradation driven by C-degron pathways
Makaros Y, Raiff A, Timms RT, Wagh AR, Gueta MI, Bekturova A, Guez-Haddad J, Brodsky S, Opatowsky Y, Glickman MH, Elledge SJ, Koren I
Although most eukaryotic proteins are targeted for proteasomal degradation by ubiquitination, a subset have been demonstrated to undergo ubiquitin-independent proteasomal degradation (UbInPD). However, little is known about the molecular mechanisms driving UbInPD and the degrons involved. Utilizing the GPS-peptidome approach, a systematic method for degron discovery, we found thousands of sequences that promote UbInPD; thus, UbInPD is more prevalent than currently appreciated. Furthermore, mutagenesis experiments revealed specific C-terminal degrons required for UbInPD. Stability profiling of a genome-wide collection of human open reading frames identified 69 full-length proteins subject to UbInPD. These included REC8 and CDCA4, proteins which control proliferation and survival, as well as mislocalized secretory proteins, suggesting that UbInPD performs both regulatory and protein quality control functions. In the context of full-length proteins, C termini also play a role in promoting UbInPD. Finally, we found that Ubiquilin family proteins mediate the proteasomal targeting of a subset of UbInPD substrates.
Although most eukaryotic proteins are targeted for proteasomal degradation by ubiquitination, a subset have been demonstrated to undergo ubiquitin-independent proteasomal degradation (UbInPD). However, little is known about the molecular mechanisms driving UbInPD and the degrons involved. Utilizing the GPS-peptidome approach, a systematic method for degron discovery, we found thousands of sequences that promote UbInPD; thus, UbInPD is more prevalent than currently appreciated. Furthermore, mutagenesis experiments revealed specific C-terminal degrons required for UbInPD. Stability profiling of a genome-wide collection of human open reading frames identified 69 full-length proteins subject to UbInPD. These included REC8 and CDCA4, proteins which control proliferation and survival, as well as mislocalized secretory proteins, suggesting that UbInPD performs both regulatory and protein quality control functions. In the context of full-length proteins, C termini also play a role in promoting UbInPD. Finally, we found that Ubiquilin family proteins mediate the proteasomal targeting of a subset of UbInPD substrates.

April 2023 | Research Article

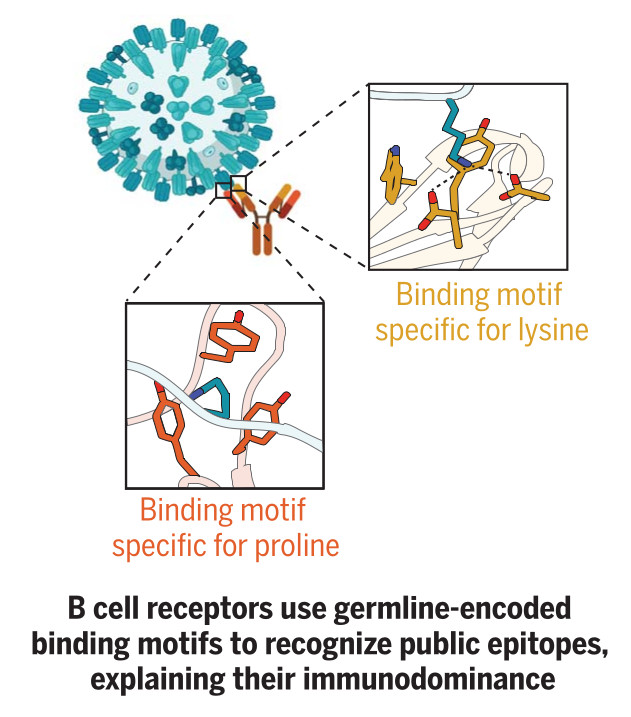
Germline-encoded amino acid-binding motifs drive immunodominant public antibody responses
Shrock EL, Timms RT, Kula T, Mena EL, West AP Jr, Guo R, Lee IH, Cohen AA, McKay LGA, Bi C, Leng Y, Fujimura E, Horns F, Li M, Wesemann DR, Griffiths A, Gewurz BE, Bjorkman PJ, Elledge SJ
Despite the vast diversity of the antibody repertoire, infected individuals often mount antibody responses to precisely the same epitopes within antigens. The immunological mechanisms underpinning this phenomenon remain unknown. By mapping 376 immunodominant "public epitopes" at high resolution and characterizing several of their cognate antibodies, we concluded that germline-encoded sequences in antibodies drive recurrent recognition. Systematic analysis of antibody-antigen structures uncovered 18 human and 21 partially overlapping mouse germline-encoded amino acid-binding (GRAB) motifs within heavy and light V gene segments that in case studies proved critical for public epitope recognition. GRAB motifs represent a fundamental component of the immune system's architecture that promotes recognition of pathogens and leads to species-specific public antibody responses that can exert selective pressure on pathogens.
Despite the vast diversity of the antibody repertoire, infected individuals often mount antibody responses to precisely the same epitopes within antigens. The immunological mechanisms underpinning this phenomenon remain unknown. By mapping 376 immunodominant "public epitopes" at high resolution and characterizing several of their cognate antibodies, we concluded that germline-encoded sequences in antibodies drive recurrent recognition. Systematic analysis of antibody-antigen structures uncovered 18 human and 21 partially overlapping mouse germline-encoded amino acid-binding (GRAB) motifs within heavy and light V gene segments that in case studies proved critical for public epitope recognition. GRAB motifs represent a fundamental component of the immune system's architecture that promotes recognition of pathogens and leads to species-specific public antibody responses that can exert selective pressure on pathogens.
January 2023 | Research Article

A central role for regulated protein stability in the control of TFE3 and MITF by nutrients
Nardone C, Palanski BA, Scott DC, Timms RT, Barber KW, Gu X, Mao A, Leng Y, Watson EV, Schulman BA, Cole PA, Elledge SJ
The TFE3 and MITF master transcription factors maintain metabolic homeostasis by regulating lysosomal, melanocytic, and autophagy genes. Previous studies posited that their cytosolic retention by 14-3-3, mediated by the Rag GTPases-mTORC1, was key for suppressing transcriptional activity in the presence of nutrients. Here, we demonstrate using mammalian cells that regulated protein stability plays a fundamental role in their control. Amino acids promote the recruitment of TFE3 and MITF to the lysosomal surface via the Rag GTPases, activating an evolutionarily conserved phospho-degron and leading to ubiquitination by CUL1β-TrCP and degradation. Elucidation of the minimal functional degron revealed a conserved alpha-helix required for interaction with RagA, illuminating the molecular basis for a severe neurodevelopmental syndrome caused by missense mutations in TFE3 within the RagA-TFE3 interface. Additionally, the phospho-degron is recurrently lost in TFE3 genomic translocations that cause kidney cancer. Therefore, two divergent pathologies converge on the loss of protein stability regulation by nutrients.
The TFE3 and MITF master transcription factors maintain metabolic homeostasis by regulating lysosomal, melanocytic, and autophagy genes. Previous studies posited that their cytosolic retention by 14-3-3, mediated by the Rag GTPases-mTORC1, was key for suppressing transcriptional activity in the presence of nutrients. Here, we demonstrate using mammalian cells that regulated protein stability plays a fundamental role in their control. Amino acids promote the recruitment of TFE3 and MITF to the lysosomal surface via the Rag GTPases, activating an evolutionarily conserved phospho-degron and leading to ubiquitination by CUL1β-TrCP and degradation. Elucidation of the minimal functional degron revealed a conserved alpha-helix required for interaction with RagA, illuminating the molecular basis for a severe neurodevelopmental syndrome caused by missense mutations in TFE3 within the RagA-TFE3 interface. Additionally, the phospho-degron is recurrently lost in TFE3 genomic translocations that cause kidney cancer. Therefore, two divergent pathologies converge on the loss of protein stability regulation by nutrients.

January 2023 | Research Article

High-throughput, targeted MHC class I immunopeptidomics using a functional genetics screening platform
Bruno PM, Timms RT, Abdelfattah NS, Leng Y, Lelis FJN, Wesemann DR, Yu XG, Elledge SJ
Identification of CD8+ T cell epitopes is critical for the development of immunotherapeutics. Existing methods for major histocompatibility complex class I (MHC class I) ligand discovery are time intensive, specialized and unable to interrogate specific proteins on a large scale. Here, we present EpiScan, which uses surface MHC class I levels as a readout for whether a genetically encoded peptide is an MHC class I ligand. Predetermined starting pools composed of >100,000 peptides can be designed using oligonucleotide synthesis, permitting large-scale MHC class I screening. We exploit this programmability of EpiScan to uncover an unappreciated role for cysteine that increases the number of predicted ligands by 9-21%, reveal affinity hierarchies by analysis of biased anchor peptide libraries and screen viral proteomes for MHC class I ligands. Using these data, we generate and iteratively refine peptide binding predictions to create EpiScan Predictor. EpiScan Predictor performs comparably to other state-of-the-art MHC class I peptide binding prediction algorithms without suffering from underrepresentation of cysteine-containing peptides. Thus, targeted immunopeptidomics using EpiScan will accelerate CD8+ T cell epitope discovery toward the goal of individual-specific immunotherapeutics.
Identification of CD8+ T cell epitopes is critical for the development of immunotherapeutics. Existing methods for major histocompatibility complex class I (MHC class I) ligand discovery are time intensive, specialized and unable to interrogate specific proteins on a large scale. Here, we present EpiScan, which uses surface MHC class I levels as a readout for whether a genetically encoded peptide is an MHC class I ligand. Predetermined starting pools composed of >100,000 peptides can be designed using oligonucleotide synthesis, permitting large-scale MHC class I screening. We exploit this programmability of EpiScan to uncover an unappreciated role for cysteine that increases the number of predicted ligands by 9-21%, reveal affinity hierarchies by analysis of biased anchor peptide libraries and screen viral proteomes for MHC class I ligands. Using these data, we generate and iteratively refine peptide binding predictions to create EpiScan Predictor. EpiScan Predictor performs comparably to other state-of-the-art MHC class I peptide binding prediction algorithms without suffering from underrepresentation of cysteine-containing peptides. Thus, targeted immunopeptidomics using EpiScan will accelerate CD8+ T cell epitope discovery toward the goal of individual-specific immunotherapeutics.

February 2021 | Research Article

Systematic characterization of mutations altering protein degradation in human cancers
Tokheim C*, Wang X*, Timms RT*, Zhang B, Mena EL, Wang B, Chen C, Ge J, Chu J, Zhang W, Elledge SJ, Brown M, Liu XS
The ubiquitin-proteasome system (UPS) is the primary route for selective protein degradation in human cells. The UPS is an attractive target for novel cancer therapies, but the precise UPS genes and substrates important for cancer growth are incompletely understood. Leveraging multi-omics data across more than 9,000 human tumors and 33 cancer types, we found that over 19% of all cancer driver genes affect UPS function. We implicate transcription factors as important substrates and show that c-Myc stability is modulated by CUL3. Moreover, we developed a deep learning model (deepDegron) to identify mutations that result in degron loss and experimentally validated the prediction that gain-of-function truncating mutations in GATA3 and PPM1D result in increased protein stability. Last, we identified UPS driver genes associated with prognosis and the tumor microenvironment. This study demonstrates the important role of UPS dysregulation in human cancer and underscores the potential therapeutic utility of targeting the UPS.
The ubiquitin-proteasome system (UPS) is the primary route for selective protein degradation in human cells. The UPS is an attractive target for novel cancer therapies, but the precise UPS genes and substrates important for cancer growth are incompletely understood. Leveraging multi-omics data across more than 9,000 human tumors and 33 cancer types, we found that over 19% of all cancer driver genes affect UPS function. We implicate transcription factors as important substrates and show that c-Myc stability is modulated by CUL3. Moreover, we developed a deep learning model (deepDegron) to identify mutations that result in degron loss and experimentally validated the prediction that gain-of-function truncating mutations in GATA3 and PPM1D result in increased protein stability. Last, we identified UPS driver genes associated with prognosis and the tumor microenvironment. This study demonstrates the important role of UPS dysregulation in human cancer and underscores the potential therapeutic utility of targeting the UPS.

October 2020 | Research Article

TASOR is a pseudo-PARP that directs HUSH complex assembly and epigenetic transposon control
Douse CH*, Tchasovnikarova IA*, Timms RT*, Protasio AV, Seczynska M, Prigozhin DM, Albecka A, Wagstaff J, Williamson JC, Freund SM, Lehner PJ, Modis Y
The HUSH complex represses retroviruses, transposons and genes to maintain the integrity of vertebrate genomes. HUSH regulates deposition of the epigenetic mark H3K9me3, but how its three core subunits — TASOR, MPP8 and Periphilin — contribute to assembly and targeting of the complex remains unknown. Here, we define the biochemical basis of HUSH assembly and find that its modular architecture resembles the yeast RNA-induced transcriptional silencing complex. TASOR, the central HUSH subunit, associates with RNA processing components. TASOR is required for H3K9me3 deposition over LINE-1 repeats and repetitive exons in transcribed genes. In the context of previous studies, this suggests that an RNA intermediate is important for HUSH activity. We dissect the TASOR and MPP8 domains necessary for transgene repression. Structure-function analyses reveal TASOR bears a catalytically-inactive PARP domain necessary for targeted H3K9me3 deposition. We conclude that TASOR is a multifunctional pseudo-PARP that directs HUSH assembly and epigenetic regulation of repetitive genomic targets.
The HUSH complex represses retroviruses, transposons and genes to maintain the integrity of vertebrate genomes. HUSH regulates deposition of the epigenetic mark H3K9me3, but how its three core subunits — TASOR, MPP8 and Periphilin — contribute to assembly and targeting of the complex remains unknown. Here, we define the biochemical basis of HUSH assembly and find that its modular architecture resembles the yeast RNA-induced transcriptional silencing complex. TASOR, the central HUSH subunit, associates with RNA processing components. TASOR is required for H3K9me3 deposition over LINE-1 repeats and repetitive exons in transcribed genes. In the context of previous studies, this suggests that an RNA intermediate is important for HUSH activity. We dissect the TASOR and MPP8 domains necessary for transgene repression. Structure-function analyses reveal TASOR bears a catalytically-inactive PARP domain necessary for targeted H3K9me3 deposition. We conclude that TASOR is a multifunctional pseudo-PARP that directs HUSH assembly and epigenetic regulation of repetitive genomic targets.

September 2020 | Research Article

Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity
Shrock E*, Fujimura E*, Kula T†, Timms RT†, Lee IH, Leng Y, Robinson ML, Sie BM, Li MZ, Chen Y, Logue J, Zuiani A, McCulloch D, Lelis FJN, Henson S, Monaco DR, Travers M, Habibi S, Clarke WA, Caturegli P, Laeyendecker O, Piechocka-Trocha A, Li J, Khatri A, Chu HY; MGH COVID-19 Collection & Processing Team, Villani AC, Kays K, Goldberg MB, Hacohen N, Filbin MR, Yu XG, Walker BD, Wesemann DR, Larman HB, Lederer JA, Elledge SJ
Understanding humoral responses to SARS-CoV-2 is critical for improving diagnostics, therapeutics, and vaccines. Deep serological profiling of 232 COVID-19 patients and 190 pre-COVID-19 era controls using VirScan revealed over 800 epitopes in the SARS-CoV-2 proteome, including 10 epitopes likely recognized by neutralizing antibodies. Pre-existing antibodies in controls recognized SARS-CoV-2 ORF1, while only COVID-19 patients primarily recognized spike and nucleoprotein. A machine learning model trained on VirScan data predicted SARS-CoV-2 exposure history with 99% sensitivity and 98% specificity; a rapid Luminex-based diagnostic was developed from the most discriminatory SARS-CoV-2 peptides. Individuals with more severe COVID-19 exhibited stronger and broader SARS-CoV-2 responses, weaker antibody responses to prior infections, and higher incidence of CMV and HSV-1, possibly influenced by demographic covariates. Among hospitalized patients, males make greater SARS-CoV-2 antibody responses than females.
Understanding humoral responses to SARS-CoV-2 is critical for improving diagnostics, therapeutics, and vaccines. Deep serological profiling of 232 COVID-19 patients and 190 pre-COVID-19 era controls using VirScan revealed over 800 epitopes in the SARS-CoV-2 proteome, including 10 epitopes likely recognized by neutralizing antibodies. Pre-existing antibodies in controls recognized SARS-CoV-2 ORF1, while only COVID-19 patients primarily recognized spike and nucleoprotein. A machine learning model trained on VirScan data predicted SARS-CoV-2 exposure history with 99% sensitivity and 98% specificity; a rapid Luminex-based diagnostic was developed from the most discriminatory SARS-CoV-2 peptides. Individuals with more severe COVID-19 exhibited stronger and broader SARS-CoV-2 responses, weaker antibody responses to prior infections, and higher incidence of CMV and HSV-1, possibly influenced by demographic covariates. Among hospitalized patients, males make greater SARS-CoV-2 antibody responses than females.

September 2020 | Research Article

Periphilin self-association underpins epigenetic silencing by the HUSH complex
Prigozhin DM, Douse CH, Farleigh LE, Albecka A, Tchasovnikarova IA, Timms RT, Oda SI, Adolf F, Freund SMV, Maslen S, Lehner PJ, Modis Y
Transcription of integrated DNA from viruses or transposable elements is tightly regulated to prevent pathogenesis. The Human Silencing Hub (HUSH), composed of Periphilin, TASOR and MPP8, silences transcriptionally active viral and endogenous transgenes. HUSH recruits effectors that alter the epigenetic landscape and chromatin structure, but how HUSH recognizes target loci and represses their expression remains unclear. We identify the physicochemical properties of Periphilin necessary for HUSH assembly and silencing. A disordered N-terminal domain (NTD) and structured C-terminal domain are essential for silencing. A crystal structure of the Periphilin-TASOR minimal core complex shows Periphilin forms an α-helical homodimer, bound by a single TASOR molecule. The NTD forms insoluble aggregates through an arginine/tyrosine-rich sequence reminiscent of low-complexity regions from self-associating RNA-binding proteins. Residues required for TASOR binding and aggregation were required for HUSH-dependent silencing and genome-wide deposition of repressive mark H3K9me3. The NTD was functionally complemented by low-complexity regions from certain RNA-binding proteins and proteins that form condensates or fibrils. Our work suggests the associative properties of Periphilin promote HUSH aggregation at target loci.
Transcription of integrated DNA from viruses or transposable elements is tightly regulated to prevent pathogenesis. The Human Silencing Hub (HUSH), composed of Periphilin, TASOR and MPP8, silences transcriptionally active viral and endogenous transgenes. HUSH recruits effectors that alter the epigenetic landscape and chromatin structure, but how HUSH recognizes target loci and represses their expression remains unclear. We identify the physicochemical properties of Periphilin necessary for HUSH assembly and silencing. A disordered N-terminal domain (NTD) and structured C-terminal domain are essential for silencing. A crystal structure of the Periphilin-TASOR minimal core complex shows Periphilin forms an α-helical homodimer, bound by a single TASOR molecule. The NTD forms insoluble aggregates through an arginine/tyrosine-rich sequence reminiscent of low-complexity regions from self-associating RNA-binding proteins. Residues required for TASOR binding and aggregation were required for HUSH-dependent silencing and genome-wide deposition of repressive mark H3K9me3. The NTD was functionally complemented by low-complexity regions from certain RNA-binding proteins and proteins that form condensates or fibrils. Our work suggests the associative properties of Periphilin promote HUSH aggregation at target loci.

July 2020 | Review Article

Tying up loose ends: the N-degron and C-degron pathways of protein degradation
Timms RT, Koren I
Selective protein degradation by the ubiquitin-proteasome system (UPS) is thought to be governed primarily by the recognition of specific motifs - degrons - present in substrate proteins. The ends of proteins - the N- and C-termini - have unique properties, and an important subset of protein-protein interactions involve the recognition of free termini. The first degrons to be discovered were located at the extreme N-terminus of proteins, a finding which initiated the study of the N-degron (formerly N-end rule) pathways, but only in the last few years has it emerged that a diverse set of C-degron pathways target analogous degron motifs located at the extreme C-terminus of proteins. In this minireview we summarise the N-degron and C-degron pathways currently known to operate in human cells, focussing primarily on those that have been discovered in recent years. In each case we describe the cellular machinery responsible for terminal degron recognition, and then consider some of the functional roles of terminal degron pathways. Altogether, a broad spectrum of E3 ubiquitin ligases mediate the recognition of a diverse array of terminal degron motifs; these degradative pathways have the potential to influence a wide variety of cellular functions.
Selective protein degradation by the ubiquitin-proteasome system (UPS) is thought to be governed primarily by the recognition of specific motifs - degrons - present in substrate proteins. The ends of proteins - the N- and C-termini - have unique properties, and an important subset of protein-protein interactions involve the recognition of free termini. The first degrons to be discovered were located at the extreme N-terminus of proteins, a finding which initiated the study of the N-degron (formerly N-end rule) pathways, but only in the last few years has it emerged that a diverse set of C-degron pathways target analogous degron motifs located at the extreme C-terminus of proteins. In this minireview we summarise the N-degron and C-degron pathways currently known to operate in human cells, focussing primarily on those that have been discovered in recent years. In each case we describe the cellular machinery responsible for terminal degron recognition, and then consider some of the functional roles of terminal degron pathways. Altogether, a broad spectrum of E3 ubiquitin ligases mediate the recognition of a diverse array of terminal degron motifs; these degradative pathways have the potential to influence a wide variety of cellular functions.

July 2019 | Research Article

A glycine-specific N-degron pathway mediates the quality control of protein N-myristoylation
Timms RT, Zhang Z, Rhee DY, Harper JW, Koren I, Elledge SJ
The N-terminal residue influences protein stability through N-degron pathways. We used stability profiling of the human N-terminome to uncover multiple additional features of N-degron pathways. In addition to uncovering extended specificities of UBR E3 ligases, we characterized two related Cullin-RING E3 ligase complexes, Cul2ZYG11B and Cul2ZER1, that act redundantly to target N-terminal glycine. N-terminal glycine degrons are depleted at native N-termini but strongly enriched at caspase cleavage sites, suggesting roles for the substrate adaptors ZYG11B and ZER1 in protein degradation during apoptosis. Furthermore, ZYG11B and ZER1 were found to participate in the quality control of N-myristoylated proteins, in which N-terminal glycine degrons are conditionally exposed after a failure of N-myristoylation. Thus, an additional N-degron pathway specific for glycine regulates the stability of metazoan proteomes.
The N-terminal residue influences protein stability through N-degron pathways. We used stability profiling of the human N-terminome to uncover multiple additional features of N-degron pathways. In addition to uncovering extended specificities of UBR E3 ligases, we characterized two related Cullin-RING E3 ligase complexes, Cul2ZYG11B and Cul2ZER1, that act redundantly to target N-terminal glycine. N-terminal glycine degrons are depleted at native N-termini but strongly enriched at caspase cleavage sites, suggesting roles for the substrate adaptors ZYG11B and ZER1 in protein degradation during apoptosis. Furthermore, ZYG11B and ZER1 were found to participate in the quality control of N-myristoylated proteins, in which N-terminal glycine degrons are conditionally exposed after a failure of N-myristoylation. Thus, an additional N-degron pathway specific for glycine regulates the stability of metazoan proteomes.

January 2019 | Protocol Article

Differential viral accessibility (DIVA) identifies alterations in chromatin architecture through large-scale mapping of lentiviral integration sites
Timms RT, Tchasovnikarova IA, Lehner PJ
Alterations in chromatin structure play a major role in the epigenetic regulation of gene expression. Here, we describe a step-by-step protocol for differential viral accessibility (DIVA), a method for identifying changes in chromatin accessibility genome-wide. Commonly used methods for mapping accessible genomic loci have strong preferences toward detecting 'open' chromatin found at regulatory regions but are not well suited to studying chromatin accessibility in gene bodies and intergenic regions. DIVA overcomes this limitation, enabling a broader range of sites to be interrogated. Conceptually, DIVA is similar to ATAC-seq in that it relies on the integration of exogenous DNA into the genome to map accessible chromatin, except that chromatin architecture is probed through mapping integration sites of exogenous lentiviruses. An isogenic pair of cell lines are transduced with a lentiviral vector, followed by PCR amplification and Illumina sequencing of virus-genome junctions; the resulting sequences define a set of unique lentiviral integration sites, which are compared to determine whether genomic loci exhibit significantly altered accessibility between experimental and control cells. Experienced researchers will take 6 d to generate lentiviral stocks and transduce the target cells, a further 5 d to prepare the Illumina sequencing libraries and a few hours to perform the bioinformatic analysis.
Alterations in chromatin structure play a major role in the epigenetic regulation of gene expression. Here, we describe a step-by-step protocol for differential viral accessibility (DIVA), a method for identifying changes in chromatin accessibility genome-wide. Commonly used methods for mapping accessible genomic loci have strong preferences toward detecting 'open' chromatin found at regulatory regions but are not well suited to studying chromatin accessibility in gene bodies and intergenic regions. DIVA overcomes this limitation, enabling a broader range of sites to be interrogated. Conceptually, DIVA is similar to ATAC-seq in that it relies on the integration of exogenous DNA into the genome to map accessible chromatin, except that chromatin architecture is probed through mapping integration sites of exogenous lentiviruses. An isogenic pair of cell lines are transduced with a lentiviral vector, followed by PCR amplification and Illumina sequencing of virus-genome junctions; the resulting sequences define a set of unique lentiviral integration sites, which are compared to determine whether genomic loci exhibit significantly altered accessibility between experimental and control cells. Experienced researchers will take 6 d to generate lentiviral stocks and transduce the target cells, a further 5 d to prepare the Illumina sequencing libraries and a few hours to perform the bioinformatic analysis.

December 2018 | Research Article

The sterol-responsive RNF145 E3 ubiquitin ligase mediates the degradation of HMG-CoA reductase together with gp78 and Hrd1
Menzies SA, Volkmar N, van den Boomen DJ, Timms RT, Dickson AS, Nathan JA, Lehner PJ
Mammalian HMG-CoA reductase (HMGCR), the rate-limiting enzyme of the cholesterol biosynthetic pathway and the therapeutic target of statins, is post-transcriptionally regulated by sterol-accelerated degradation. Under cholesterol-replete conditions, HMGCR is ubiquitinated and degraded, but the identity of the E3 ubiquitin ligase(s) responsible for mammalian HMGCR turnover remains controversial. Using systematic, unbiased CRISPR/Cas9 genome-wide screens with a sterol-sensitive endogenous HMGCR reporter, we comprehensively map the E3 ligase landscape required for sterol-accelerated HMGCR degradation. We find that RNF145 and gp78 independently co-ordinate HMGCR ubiquitination and degradation. RNF145, a sterol-responsive ER-resident E3 ligase, is unstable but accumulates following sterol depletion. Sterol addition triggers RNF145 recruitment to HMGCR via Insigs, promoting HMGCR ubiquitination and proteasome-mediated degradation. In the absence of both RNF145 and gp78, Hrd1, a third UBE2G2-dependent E3 ligase, partially regulates HMGCR activity. Our findings reveal a critical role for the sterol-responsive RNF145 in HMGCR regulation and elucidate the complexity of sterol-accelerated HMGCR degradation.
Mammalian HMG-CoA reductase (HMGCR), the rate-limiting enzyme of the cholesterol biosynthetic pathway and the therapeutic target of statins, is post-transcriptionally regulated by sterol-accelerated degradation. Under cholesterol-replete conditions, HMGCR is ubiquitinated and degraded, but the identity of the E3 ubiquitin ligase(s) responsible for mammalian HMGCR turnover remains controversial. Using systematic, unbiased CRISPR/Cas9 genome-wide screens with a sterol-sensitive endogenous HMGCR reporter, we comprehensively map the E3 ligase landscape required for sterol-accelerated HMGCR degradation. We find that RNF145 and gp78 independently co-ordinate HMGCR ubiquitination and degradation. RNF145, a sterol-responsive ER-resident E3 ligase, is unstable but accumulates following sterol depletion. Sterol addition triggers RNF145 recruitment to HMGCR via Insigs, promoting HMGCR ubiquitination and proteasome-mediated degradation. In the absence of both RNF145 and gp78, Hrd1, a third UBE2G2-dependent E3 ligase, partially regulates HMGCR activity. Our findings reveal a critical role for the sterol-responsive RNF145 in HMGCR regulation and elucidate the complexity of sterol-accelerated HMGCR degradation.

June 2018 | Research Article

The Eukaryotic Proteome Is Shaped by E3 Ubiquitin Ligases Targeting C-Terminal Degrons
Koren I, Timms RT, Kula T, Xu Q, Li MZ, Elledge SJ
Degrons are minimal elements that mediate the interaction of proteins with degradation machineries to promote proteolysis. Despite their central role in proteostasis, the number of known degrons remains small, and a facile technology to characterize them is lacking. Using a strategy combining global protein stability (GPS) profiling with a synthetic human peptidome, we identify thousands of peptides containing degron activity. Employing CRISPR screening, we establish that the stability of many proteins is regulated through degrons located at their C terminus. We characterize eight Cullin-RING E3 ubiquitin ligase (CRL) complex adaptors that regulate C-terminal degrons, including six CRL2 and two CRL4 complexes, and computationally implicate multiple non-CRLs in end recognition. Proteome analysis revealed that the C termini of eukaryotic proteins are depleted for C-terminal degrons, suggesting an E3-ligase-dependent modulation of proteome composition. Thus, we propose that a series of "C-end rules" operate to govern protein stability and shape the eukaryotic proteome.
Degrons are minimal elements that mediate the interaction of proteins with degradation machineries to promote proteolysis. Despite their central role in proteostasis, the number of known degrons remains small, and a facile technology to characterize them is lacking. Using a strategy combining global protein stability (GPS) profiling with a synthetic human peptidome, we identify thousands of peptides containing degron activity. Employing CRISPR screening, we establish that the stability of many proteins is regulated through degrons located at their C terminus. We characterize eight Cullin-RING E3 ubiquitin ligase (CRL) complex adaptors that regulate C-terminal degrons, including six CRL2 and two CRL4 complexes, and computationally implicate multiple non-CRLs in end recognition. Proteome analysis revealed that the C termini of eukaryotic proteins are depleted for C-terminal degrons, suggesting an E3-ligase-dependent modulation of proteome composition. Thus, we propose that a series of "C-end rules" operate to govern protein stability and shape the eukaryotic proteome.

May 2018 | Research Article

The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes
Robbez-Masson L, Tie CHC, Conde L, Tunbak H, Husovsky C, Tchasovnikarova IA, Timms RT, Herrero J, Lehner PJ, Rowe HM
Retrotransposons encompass half of the human genome and contribute to the formation of heterochromatin, which provides nuclear structure and regulates gene expression. Here, we asked if the human silencing hub (HUSH) complex is necessary to silence retrotransposons and whether it collaborates with TRIM28 and the chromatin remodeler ATRX at specific genomic loci. We show that the HUSH complex contributes to de novo repression and DNA methylation of an SVA retrotransposon reporter. By using naïve versus primed mouse pluripotent stem cells, we reveal a critical role for the HUSH complex in naïve cells, implicating it in programming epigenetic marks in development. Although the HUSH component FAM208A binds to endogenous retroviruses (ERVs) and long interspersed element-1s (LINE-1s or L1s), it is mainly required to repress evolutionarily young L1s (mouse-specific lineages <5 million years old). TRIM28, in contrast, is necessary to repress both ERVs and young L1s. Genes co-repressed by TRIM28 and FAM208A are evolutionarily young, or exhibit tissue-specific expression, are enriched in young L1s, and display evidence for regulation through LTR promoters. Finally, we demonstrate that the HUSH complex is also required to repress L1 elements in human cells. Overall, these data indicate that the HUSH complex and TRIM28 co-repress young retrotransposons and new genes rewired by retrotransposon noncoding DNA.
Retrotransposons encompass half of the human genome and contribute to the formation of heterochromatin, which provides nuclear structure and regulates gene expression. Here, we asked if the human silencing hub (HUSH) complex is necessary to silence retrotransposons and whether it collaborates with TRIM28 and the chromatin remodeler ATRX at specific genomic loci. We show that the HUSH complex contributes to de novo repression and DNA methylation of an SVA retrotransposon reporter. By using naïve versus primed mouse pluripotent stem cells, we reveal a critical role for the HUSH complex in naïve cells, implicating it in programming epigenetic marks in development. Although the HUSH component FAM208A binds to endogenous retroviruses (ERVs) and long interspersed element-1s (LINE-1s or L1s), it is mainly required to repress evolutionarily young L1s (mouse-specific lineages <5 million years old). TRIM28, in contrast, is necessary to repress both ERVs and young L1s. Genes co-repressed by TRIM28 and FAM208A are evolutionarily young, or exhibit tissue-specific expression, are enriched in young L1s, and display evidence for regulation through LTR promoters. Finally, we demonstrate that the HUSH complex is also required to repress L1 elements in human cells. Overall, these data indicate that the HUSH complex and TRIM28 co-repress young retrotransposons and new genes rewired by retrotransposon noncoding DNA.

February 2018 | Research Article

Neuropathic MORC2 mutations perturb GHKL ATPase dimerization dynamics and epigenetic silencing by multiple structural mechanisms
Douse CH, Bloor S, Liu Y, Shamin M, Tchasovnikarova IA, Timms RT, Lehner PJ, Modis Y
Missense mutations in MORC2 cause neuropathies including spinal muscular atrophy and Charcot-Marie-Tooth disease. We recently identified MORC2 as an effector of epigenetic silencing by the human silencing hub (HUSH). Here we report the biochemical and cellular activities of MORC2 variants, alongside crystal structures of wild-type and neuropathic forms of a human MORC2 fragment comprising the GHKL-type ATPase module and CW-type zinc finger. This fragment dimerizes upon binding ATP and contains a hinged, functionally critical coiled-coil insertion absent in other GHKL ATPases. We find that dimerization and DNA binding of the MORC2 ATPase module transduce HUSH-dependent silencing. Disease mutations change the dynamics of dimerization by distinct structural mechanisms: destabilizing the ATPase-CW module, trapping the ATP lid, or perturbing the dimer interface. These defects lead to the modulation of HUSH function, thus providing a molecular basis for understanding MORC2-associated neuropathies.
Missense mutations in MORC2 cause neuropathies including spinal muscular atrophy and Charcot-Marie-Tooth disease. We recently identified MORC2 as an effector of epigenetic silencing by the human silencing hub (HUSH). Here we report the biochemical and cellular activities of MORC2 variants, alongside crystal structures of wild-type and neuropathic forms of a human MORC2 fragment comprising the GHKL-type ATPase module and CW-type zinc finger. This fragment dimerizes upon binding ATP and contains a hinged, functionally critical coiled-coil insertion absent in other GHKL ATPases. We find that dimerization and DNA binding of the MORC2 ATPase module transduce HUSH-dependent silencing. Disease mutations change the dynamics of dimerization by distinct structural mechanisms: destabilizing the ATPase-CW module, trapping the ATP lid, or perturbing the dimer interface. These defects lead to the modulation of HUSH function, thus providing a molecular basis for understanding MORC2-associated neuropathies.

June 2017 | Research Article

Hyperactivation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2
Tchasovnikarova IA*, Timms RT*, Douse CH, Roberts RC, Dougan G, Kingston RE, Modis Y, Lehner P
Dominant mutations in the MORC2 gene have recently been shown to cause axonal Charcot-Marie-Tooth (CMT) disease, but the cellular function of MORC2 is poorly understood. Here, through a genome-wide CRISPR-Cas9-mediated forward genetic screen, we identified MORC2 as an essential gene required for epigenetic silencing by the HUSH complex. HUSH recruits MORC2 to target sites in heterochromatin. We exploited a new method, differential viral accessibility (DIVA), to show that loss of MORC2 results in chromatin decompaction at these target loci, which is concomitant with a loss of H3K9me3 deposition and transcriptional derepression. The ATPase activity of MORC2 is critical for HUSH-mediated silencing, and the most common alteration affecting the ATPase domain in CMT patients (p.Arg252Trp) hyperactivates HUSH-mediated repression in neuronal cells. These data define a critical role for MORC2 in epigenetic silencing by the HUSH complex and provide a mechanistic basis underpinning the role of MORC2 mutations in CMT disease.
Dominant mutations in the MORC2 gene have recently been shown to cause axonal Charcot-Marie-Tooth (CMT) disease, but the cellular function of MORC2 is poorly understood. Here, through a genome-wide CRISPR-Cas9-mediated forward genetic screen, we identified MORC2 as an essential gene required for epigenetic silencing by the HUSH complex. HUSH recruits MORC2 to target sites in heterochromatin. We exploited a new method, differential viral accessibility (DIVA), to show that loss of MORC2 results in chromatin decompaction at these target loci, which is concomitant with a loss of H3K9me3 deposition and transcriptional derepression. The ATPase activity of MORC2 is critical for HUSH-mediated silencing, and the most common alteration affecting the ATPase domain in CMT patients (p.Arg252Trp) hyperactivates HUSH-mediated repression in neuronal cells. These data define a critical role for MORC2 in epigenetic silencing by the HUSH complex and provide a mechanistic basis underpinning the role of MORC2 mutations in CMT disease.

October 2016 | Research Article

Mitochondrial Protein Lipoylation and the 2-Oxoglutarate Dehydrogenase Complex Controls HIF1α Stability in Aerobic Conditions
Burr SP, Costa AS, Grice GL, Timms RT, Lobb IT, Freisinger P, Dodd RB, Dougan G, Lehner PJ, Frezza C, Nathan JA
Hypoxia-inducible transcription factors (HIFs) control adaptation to low oxygen environments by activating genes involved in metabolism, angiogenesis, and redox homeostasis. The finding that HIFs are also regulated by small molecule metabolites highlights the need to understand the complexity of their cellular regulation. Here we use a forward genetic screen in near-haploid human cells to identify genes that stabilize HIFs under aerobic conditions. We identify two mitochondrial genes, oxoglutarate dehydrogenase (OGDH) and lipoic acid synthase (LIAS), which when mutated stabilize HIF1α in a non-hydroxylated form. Disruption of OGDH complex activity in OGDH or LIAS mutants promotes L-2-hydroxyglutarate formation, which inhibits the activity of the HIFα prolyl hydroxylases (PHDs) and TET 2-oxoglutarate dependent dioxygenases. We also find that PHD activity is decreased in patients with homozygous germline mutations in lipoic acid synthesis, leading to HIF1 activation. Thus, mutations affecting OGDHC activity may have broad implications for epigenetic regulation and tumorigenesis.
Hypoxia-inducible transcription factors (HIFs) control adaptation to low oxygen environments by activating genes involved in metabolism, angiogenesis, and redox homeostasis. The finding that HIFs are also regulated by small molecule metabolites highlights the need to understand the complexity of their cellular regulation. Here we use a forward genetic screen in near-haploid human cells to identify genes that stabilize HIFs under aerobic conditions. We identify two mitochondrial genes, oxoglutarate dehydrogenase (OGDH) and lipoic acid synthase (LIAS), which when mutated stabilize HIF1α in a non-hydroxylated form. Disruption of OGDH complex activity in OGDH or LIAS mutants promotes L-2-hydroxyglutarate formation, which inhibits the activity of the HIFα prolyl hydroxylases (PHDs) and TET 2-oxoglutarate dependent dioxygenases. We also find that PHD activity is decreased in patients with homozygous germline mutations in lipoic acid synthesis, leading to HIF1 activation. Thus, mutations affecting OGDHC activity may have broad implications for epigenetic regulation and tumorigenesis.

October 2016 | Research Article

ATF7IP-Mediated Stabilization of the Histone Methyltransferase SETDB1 Is Essential for Heterochromatin Formation by the HUSH Complex
Timms RT*, Tchasovnikarova IA*, Antrobus R, Dougan G, Lehner PJ
The histone methyltransferase SETDB1 plays a central role in repressive chromatin processes, but the functional requirement for its binding partner ATF7IP has remained enigmatic. Here, we show that ATF7IP is essential for SETDB1 stability: nuclear SETDB1 protein is degraded by the proteasome upon ablation of ATF7IP. As a result, ATF7IP is critical for repression that requires H3K9 trimethylation by SETDB1, including transgene silencing by the HUSH complex. Furthermore, we show that loss of ATF7IP phenocopies loss of SETDB1 in genome-wide assays. ATF7IP and SETDB1 knockout cells exhibit near-identical defects in the global deposition of H3K9me3, which results in similar dysregulation of the transcriptome. Overall, these data identify a critical functional role for ATF7IP in heterochromatin formation by regulating SETDB1 abundance in the nucleus.
The histone methyltransferase SETDB1 plays a central role in repressive chromatin processes, but the functional requirement for its binding partner ATF7IP has remained enigmatic. Here, we show that ATF7IP is essential for SETDB1 stability: nuclear SETDB1 protein is degraded by the proteasome upon ablation of ATF7IP. As a result, ATF7IP is critical for repression that requires H3K9 trimethylation by SETDB1, including transgene silencing by the HUSH complex. Furthermore, we show that loss of ATF7IP phenocopies loss of SETDB1 in genome-wide assays. ATF7IP and SETDB1 knockout cells exhibit near-identical defects in the global deposition of H3K9me3, which results in similar dysregulation of the transcriptome. Overall, these data identify a critical functional role for ATF7IP in heterochromatin formation by regulating SETDB1 abundance in the nucleus.

July 2016 | Research Article

A Genetic Screen Identifies a Critical Role for the WDR81-WDR91 Complex in the Trafficking and Degradation of Tetherin
Rapiteanu R, Davis LJ, Williamson JC, Timms RT, Paul Luzio J, Lehner PJ
Tetherin (BST2/CD317) is a viral restriction factor that anchors enveloped viruses to host cells and limits viral spread. The HIV-1 Vpu accessory protein counteracts tetherin by decreasing its cell surface expression and targeting it for ubiquitin-dependent endolysosomal degradation. Although the Vpu-mediated downregulation of tetherin has been extensively studied, the molecular details are not completely elucidated. We therefore used a forward genetic screen in human haploid KBM7 cells to identify novel genes required for tetherin trafficking. Our screen identified WDR81 as a novel gene required for tetherin trafficking and degradation in both the presence and absence of Vpu. WDR81 is a BEACH-domain containing protein that is also required for the degradation of EGF-stimulated epidermal growth factor receptor (EGFR) and functions in a complex with the WDR91 protein. In the absence of WDR81 the endolysosomal compartment appears swollen, with enlarged early and late endosomes and reduced delivery of endocytosed dextran to cathepsin-active lysosomes. Our data suggest a role for the WDR81-WDR91 complex in the fusion of endolysosomal compartments and the absence of WDR81 leads to impaired receptor trafficking and degradation.
Tetherin (BST2/CD317) is a viral restriction factor that anchors enveloped viruses to host cells and limits viral spread. The HIV-1 Vpu accessory protein counteracts tetherin by decreasing its cell surface expression and targeting it for ubiquitin-dependent endolysosomal degradation. Although the Vpu-mediated downregulation of tetherin has been extensively studied, the molecular details are not completely elucidated. We therefore used a forward genetic screen in human haploid KBM7 cells to identify novel genes required for tetherin trafficking. Our screen identified WDR81 as a novel gene required for tetherin trafficking and degradation in both the presence and absence of Vpu. WDR81 is a BEACH-domain containing protein that is also required for the degradation of EGF-stimulated epidermal growth factor receptor (EGFR) and functions in a complex with the WDR91 protein. In the absence of WDR81 the endolysosomal compartment appears swollen, with enlarged early and late endosomes and reduced delivery of endocytosed dextran to cathepsin-active lysosomes. Our data suggest a role for the WDR81-WDR91 complex in the fusion of endolysosomal compartments and the absence of WDR81 leads to impaired receptor trafficking and degradation.

June 2016 | Research Article

Genetic dissection of mammalian ERAD through comparative haploid and CRISPR forward genetic screens
Timms RT*, Menzies SA*, Tchasovnikarova IA*, Christensen LC*, Williamson JC, Antrobus R, Dougan G, Ellgaard L, Lehner PJ
The application of forward genetic screens to cultured human cells represents a powerful method to study gene function. The repurposing of the bacterial CRISPR/Cas9 system provides an effective method to disrupt gene function in mammalian cells, and has been applied to genome-wide screens. Here, we compare the efficacy of genome-wide CRISPR/Cas9-mediated forward genetic screens versus gene-trap mutagenesis screens in haploid human cells, which represent the existing 'gold standard' method. This head-to-head comparison aimed to identify genes required for the endoplasmic reticulum-associated degradation (ERAD) of MHC class I molecules. The two approaches show high concordance (>70%), successfully identifying the majority of the known components of the canonical glycoprotein ERAD pathway. Both screens also identify a role for the uncharacterized gene TXNDC11, which we show encodes an EDEM2/3-associated disulphide reductase. Genome-wide CRISPR/Cas9-mediated screens together with haploid genetic screens provide a powerful addition to the forward genetic toolbox.
The application of forward genetic screens to cultured human cells represents a powerful method to study gene function. The repurposing of the bacterial CRISPR/Cas9 system provides an effective method to disrupt gene function in mammalian cells, and has been applied to genome-wide screens. Here, we compare the efficacy of genome-wide CRISPR/Cas9-mediated forward genetic screens versus gene-trap mutagenesis screens in haploid human cells, which represent the existing 'gold standard' method. This head-to-head comparison aimed to identify genes required for the endoplasmic reticulum-associated degradation (ERAD) of MHC class I molecules. The two approaches show high concordance (>70%), successfully identifying the majority of the known components of the canonical glycoprotein ERAD pathway. Both screens also identify a role for the uncharacterized gene TXNDC11, which we show encodes an EDEM2/3-associated disulphide reductase. Genome-wide CRISPR/Cas9-mediated screens together with haploid genetic screens provide a powerful addition to the forward genetic toolbox.

February 2016 | Review Article

Position-effect variegation revisited: HUSHing up heterochromatin in human cells
Timms RT, Tchasovnikarova IA, Lehner PJ
Much of what we understand about heterochromatin formation in mammals has been extrapolated from forward genetic screens for modifiers of position-effect variegation (PEV) in the fruit fly Drosophila melanogaster. The recent identification of the HUSH (Human Silencing Hub) complex suggests that more recent evolutionary developments contribute to the mechanisms underlying PEV in human cells. Although HUSH-mediated repression also involves heterochromatin spreading through the reading and writing of the repressive H3K9me3 histone modification, clear orthologues of HUSH subunits are not found in Drosophila but are conserved in vertebrates. Here we compare the insights into the mechanisms of PEV derived from genetic screens in the fly, the mouse and in human cells, review what is currently known about the HUSH complex and discuss the implications of HUSH-mediated silencing for viral latency. Future studies will provide mechanistic insight into HUSH complex function and reveal the relationship between HUSH and other epigenetic silencing complexes.
Much of what we understand about heterochromatin formation in mammals has been extrapolated from forward genetic screens for modifiers of position-effect variegation (PEV) in the fruit fly Drosophila melanogaster. The recent identification of the HUSH (Human Silencing Hub) complex suggests that more recent evolutionary developments contribute to the mechanisms underlying PEV in human cells. Although HUSH-mediated repression also involves heterochromatin spreading through the reading and writing of the repressive H3K9me3 histone modification, clear orthologues of HUSH subunits are not found in Drosophila but are conserved in vertebrates. Here we compare the insights into the mechanisms of PEV derived from genetic screens in the fly, the mouse and in human cells, review what is currently known about the HUSH complex and discuss the implications of HUSH-mediated silencing for viral latency. Future studies will provide mechanistic insight into HUSH complex function and reveal the relationship between HUSH and other epigenetic silencing complexes.

June 2015 | Research Article

Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells
Tchasovnikarova IA*, Timms RT*, Matheson NJ, Wals K, Antrobus R, Göttgens B, Dougan G, Dawson MA, Lehner PJ
Forward genetic screens in Drosophila melanogaster for modifiers of position-effect variegation have revealed the basis of much of our understanding of heterochromatin. We took an analogous approach to identify genes required for epigenetic repression in human cells. A nonlethal forward genetic screen in near-haploid KBM7 cells identified the HUSH (human silencing hub) complex, comprising three poorly characterized proteins, TASOR, MPP8, and periphilin; this complex is absent from Drosophila but is conserved from fish to humans. Loss of HUSH components resulted in decreased H3K9me3 both at endogenous genomic loci and at retroviruses integrated into heterochromatin. Our results suggest that the HUSH complex is recruited to genomic loci rich in H3K9me3, where subsequent recruitment of the methyltransferase SETDB1 is required for further H3K9me3 deposition to maintain transcriptional silencing.
Forward genetic screens in Drosophila melanogaster for modifiers of position-effect variegation have revealed the basis of much of our understanding of heterochromatin. We took an analogous approach to identify genes required for epigenetic repression in human cells. A nonlethal forward genetic screen in near-haploid KBM7 cells identified the HUSH (human silencing hub) complex, comprising three poorly characterized proteins, TASOR, MPP8, and periphilin; this complex is absent from Drosophila but is conserved from fish to humans. Loss of HUSH components resulted in decreased H3K9me3 both at endogenous genomic loci and at retroviruses integrated into heterochromatin. Our results suggest that the HUSH complex is recruited to genomic loci rich in H3K9me3, where subsequent recruitment of the methyltransferase SETDB1 is required for further H3K9me3 deposition to maintain transcriptional silencing.

July 2014 | Research Article

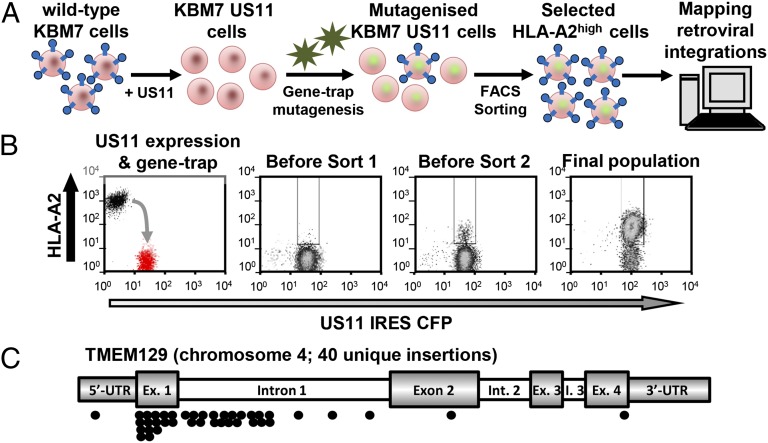
TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I
van den Boomen DJ, Timms RT, Grice GL, Stagg HR, Skødt K, Dougan G, Nathan JA, Lehner PJ
The US11 gene product of human cytomegalovirus promotes viral immune evasion by hijacking the endoplasmic reticulum (ER)-associated degradation (ERAD) pathway. US11 initiates dislocation of newly translocated MHC I from the ER to the cytosol for proteasome-mediated degradation. Despite the critical role for ubiquitin in this degradation pathway, the responsible E3 ligase is unknown. In a forward genetic screen for host ERAD components hijacked by US11 in near-haploid KBM7 cells, we identified TMEM129, an uncharacterized polytopic membrane protein. TMEM129 is essential and rate-limiting for US11-mediated MHC-I degradation and acts as a novel ER resident E3 ubiquitin ligase. TMEM129 contains an unusual cysteine-only RING with intrinsic E3 ligase activity and is recruited to US11 via Derlin-1. Together with its E2 conjugase Ube2J2, TMEM129 is responsible for the ubiquitination, dislocation, and subsequent degradation of US11-associated MHC-I. US11 engages two degradation pathways: a Derlin-1/TMEM129-dependent pathway required for MHC-I degradation and a SEL1L/HRD1-dependent pathway required for "free" US11 degradation. Our data show that TMEM129 is a novel ERAD E3 ligase and the central component of a novel mammalian ERAD complex.
The US11 gene product of human cytomegalovirus promotes viral immune evasion by hijacking the endoplasmic reticulum (ER)-associated degradation (ERAD) pathway. US11 initiates dislocation of newly translocated MHC I from the ER to the cytosol for proteasome-mediated degradation. Despite the critical role for ubiquitin in this degradation pathway, the responsible E3 ligase is unknown. In a forward genetic screen for host ERAD components hijacked by US11 in near-haploid KBM7 cells, we identified TMEM129, an uncharacterized polytopic membrane protein. TMEM129 is essential and rate-limiting for US11-mediated MHC-I degradation and acts as a novel ER resident E3 ubiquitin ligase. TMEM129 contains an unusual cysteine-only RING with intrinsic E3 ligase activity and is recruited to US11 via Derlin-1. Together with its E2 conjugase Ube2J2, TMEM129 is responsible for the ubiquitination, dislocation, and subsequent degradation of US11-associated MHC-I. US11 engages two degradation pathways: a Derlin-1/TMEM129-dependent pathway required for MHC-I degradation and a SEL1L/HRD1-dependent pathway required for "free" US11 degradation. Our data show that TMEM129 is a novel ERAD E3 ligase and the central component of a novel mammalian ERAD complex.
November 2013 | Research Article

Haploid genetic screens identify an essential role for PLP2 in the downregulation of novel plasma membrane targets by viral E3 ubiquitin ligases
Timms RT, Duncan LM, Tchasovnikarova IA, Antrobus R, Smith DL, Dougan G, Weekes MP, Lehner PJ
The Kaposi's sarcoma-associated herpesvirus gene products K3 and K5 are viral ubiquitin E3 ligases which downregulate MHC-I and additional cell surface immunoreceptors. To identify novel cellular genes required for K5 function we performed a forward genetic screen in near-haploid human KBM7 cells. The screen identified proteolipid protein 2 (PLP2), a MARVEL domain protein of unknown function, as essential for K5 activity. Genetic loss of PLP2 traps the viral ligase in the endoplasmic reticulum, where it is unable to ubiquitinate and degrade its substrates. Subsequent analysis of the plasma membrane proteome of K5-expressing KBM7 cells in the presence and absence of PLP2 revealed a wide range of novel K5 targets, all of which required PLP2 for their K5-mediated downregulation. This work ascribes a critical function to PLP2 for viral ligase activity and underlines the power of non-lethal haploid genetic screens in human cells to identify the genes involved in pathogen manipulation of the host immune system.
The Kaposi's sarcoma-associated herpesvirus gene products K3 and K5 are viral ubiquitin E3 ligases which downregulate MHC-I and additional cell surface immunoreceptors. To identify novel cellular genes required for K5 function we performed a forward genetic screen in near-haploid human KBM7 cells. The screen identified proteolipid protein 2 (PLP2), a MARVEL domain protein of unknown function, as essential for K5 activity. Genetic loss of PLP2 traps the viral ligase in the endoplasmic reticulum, where it is unable to ubiquitinate and degrade its substrates. Subsequent analysis of the plasma membrane proteome of K5-expressing KBM7 cells in the presence and absence of PLP2 revealed a wide range of novel K5 targets, all of which required PLP2 for their K5-mediated downregulation. This work ascribes a critical function to PLP2 for viral ligase activity and underlines the power of non-lethal haploid genetic screens in human cells to identify the genes involved in pathogen manipulation of the host immune system.

June 2012 | Research Article

Fluorescence-based phenotypic selection allows forward genetic screens in haploid human cells
Duncan LM*, Timms RT*, Zavodszky E, Cano F, Dougan G, Randow F, Lehner PJ
The isolation of haploid cell lines has recently allowed the power of forward genetic screens to be applied to mammalian cells. The interest in applying this powerful genetic approach to a mammalian system is only tempered by the limited utility of these screens, if confined to lethal phenotypes. Here we expand the scope of these approaches beyond live/dead screens and show that selection for a cell surface phenotype via fluorescence-activated cell sorting can identify the key molecules in an intracellular pathway, in this case MHC class I antigen presentation. Non-lethal haploid genetic screens are widely applicable to identify genes involved in essentially any cellular pathway.
The isolation of haploid cell lines has recently allowed the power of forward genetic screens to be applied to mammalian cells. The interest in applying this powerful genetic approach to a mammalian system is only tempered by the limited utility of these screens, if confined to lethal phenotypes. Here we expand the scope of these approaches beyond live/dead screens and show that selection for a cell surface phenotype via fluorescence-activated cell sorting can identify the key molecules in an intracellular pathway, in this case MHC class I antigen presentation. Non-lethal haploid genetic screens are widely applicable to identify genes involved in essentially any cellular pathway.
